

Мукополісахаридози - це цілий ряд генетично детермінованих (обумовлених) патологій, що з'являються при порушенні обміну кислихмукополісахаридів (глікозаміногліканів).
Найчастіше при такому порушенні уражається скелет і затримується фізичний розвиток, а деякі різновиди патології проявляються відсталістю з боку інтелектуальної сфери. Крім цього, спостерігаються збої в роботі серцево-судинної, шлунково-кишкової та нервової систем, зорового апарату і шкіри.
Лікування мукополисахаридозов симптоматичне, прогноз в цілому несприятливий - порушення прогресують, тому що глюкозаміноглікани накопичуються. Пацієнти вмирають досить рано.
Загальні дані
Мукополісахарідоз вивчили відносно недавно - в 20 столітті. Спершу він розцінювався як одна патологія, потім в силу різноманітності стали виділяти його типи. Об'єднуючим фактором всіх мукополисахаридозов є те, що в органах і тканинах накопичуються кислі мукополісахариди, а причиною такого порушення є неповноцінність деяких клітинних ферментів, яка закладена в генотипі і передається у спадок.
Зверніть увагуЛікування пацієнтів з мукополісахаридозом проводять ортопеди, але при цьому також потрібні клінічні зусилля кардіологів, офтальмологів, неврологів, отоларингологів і деяких інших фахівців.
Мукополісахарідоз типу IH
Мукополісахарідоз типу IH ще називають синдромом Гурлера. Він діагностується у 1 новонародженого на 20-25 тисяч.
Перші ознаки захворювання починають з'являтися протягом першого року життя дитини, але повна клінічна картина може розвинутися тільки до 2 років.
Найбільш типовими симптомами хвороби є:
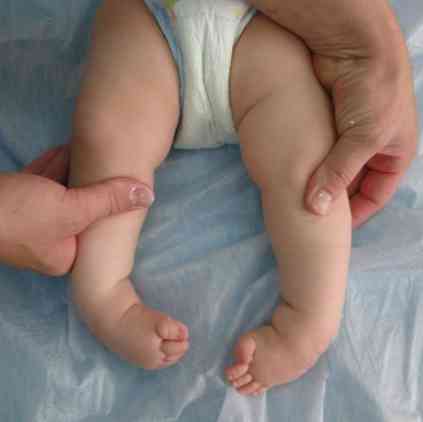 грубі риси обличчя - дуже типові для даної патології;
грубі риси обличчя - дуже типові для даної патології;- деформація черепа - він стає схожий на човновий кіль. Такий стан називається скароцефаліей;
- дихання здійснюється через рот, так як носове дихання порушене через збільшення аденоїдів, а також аномалій внутрішньоутробного розвитку в області лицьового черепа;
- деформація кінцівок і деяких інших фрагментів скелета;
- відставання в рості;
- викривлення хребта (симптом "котячої спини" в сидячому положенні);
- вкорочення шиї;
- нетипове високе розташування лопаток;
- випинання вперед нижніх ребер;
- збільшення ширини кистей - вони при цьому нагадують лапу з пазурами;
- розвиток контрактур (зниження обсягу рухів) з поступовим втягуванням в патологічний процес різних суглобів. Спершу страждають ліктьові і плечові суглоби, далі процес поширюється на колінні, тазостегнові і гомілковостопні зчленування.
 грубі риси обличчя - дуже типові для даної патології;
грубі риси обличчя - дуже типові для даної патології;Для синдрому Гурлера характерна специфічна хода - навшпиньки з напівзігнутими ногами. Така особливість виникає через обмеження рухів через контрактур.
При фізикальному обстеженні виявляється наступне:
- живіт збільшений, його передня стінка у таких хворих ослаблена, тому нерідко можуть розвиватися пупкові і пахові грижі;
- печінка і селезінка збільшені;
- у чоловіків часто виявляється гидроцеле - водянка яєчка;
- в ряді випадків виявляються порушення координації рухів;
- виявляється надмірний ріст Пушкова волосся.
При інструментальному обстеженні визначається наступне:
- на електрокардіограмі - ознаки дифузного (поширеного) ураження серцевого м'яза;
- порушення з боку зору та слуху;
- при офтальмоскопії виявляють помутніння і збільшення рогівки.
Найбільш характерними є зміни, які констатують при проведенні рентгенологічного обстеження:
- рентгенографія хребта - дозволяє виявити характерну деформацію хребта. При цьому хребці мають форму куба, їх краї закруглені. Також відзначається кіфоз - викривлення хребта опуклістю вперед;
- рентгенографія грудної клітки - наголошується потовщення передніх фрагментів ребер з одночасним витончення їх задніх відділів, укороченням і деформацією. Ребра при цьому набувають специфічний вид і стають схожими на лопату або шаблю. Також відзначається зменшення і деякий зсув головок плечових кісток;
- рентгенографія тазу - на ній виявляється звуження тазового кільця, вертлюжної западини (ті, в які є "основою" формування тазостегнового суглоба) скошені, головки стегнових кісток зменшені;
- рентгенографія трубчастих кісток - відзначається стоншення кортикального (поверхневого) шару кісток і деяке розширення кістковомозкового каналу;
- рентгенографія кісток кисті - допомагає виявити недорозвинення дистальних (нігтьових) фаланг пальців, вкорочення і розширення середніх і проксимальних (тих, які знаходяться ближче до долоні) фаланг, а також п'ясткових кісток;
- рентгенографія черепа - чітко виявляється недорозвиненість кісток лицьового черепа, краніостеноз (звуження черепа в деяких локаціях) і макроцефалія (збільшення черепа).
У хворих з діагнозом мукополисахаридоза типу IH виявляються такі порушення, які можуть розцінюватися як окремі хвороби:
- пігментна дистрофія сітківки - порушення її розвитку;
- глаукома - підвищення внутрішньоочного тиску;
- застійні явища в області очного дна;
- парези - порушення руху;
- паралічі - повне порушення рухової активності в певних ділянках опорно-рухового апарату.
Згодом у пацієнтів з діагнозом Гурлера розвивається і необоротно прогресує розумова відсталість.
Мукополісахарідоз типу I-S
Інша назва мукополисахаридоза типу I-S - хвороба Шейе. Захворювання було описано тільки в середині 20 століття, хоча пацієнти з характерними для нього контрактурами пальців кистей, порушеннями рухів у великих суглобах і специфічними рисами обличчя зустрічалися в практиці клініцистів і раніше. Ознаки можуть комбінуватися з ознаками синдрому Гурлера.
Цей вид мукополисахаридоза з'являється в більш пізньому віці, ніж при мукополісахарідозов типу IH, а його протягом більш сприятливе.
Характерною особливістю мукополисахаридоза типу I-S є те, що до 3-6 років життя розвиток дітей відповідає віковій нормі. Далі починають проявлятися такі ознаки:
- згинальні контрактури пальців рук;
- обмеження розгинання в великих і середніх суглобах - а саме ліктьових, плечових і променезап'ясткових.
Порушення амплітуди рухів з боку нижніх кінцівок менш виражене, ніж при інших видах описуваної патології.
Повноцінна клінічна картина цього виду мукополисахаридоза встановлюється до початку підліткового періоду.
При фізіологічному дослідженні (огляді) відзначається наступне:
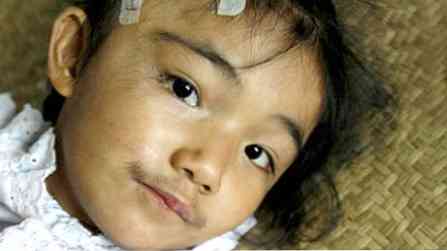 хворі невисокі, але мають міцну статуру (кремезні);
хворі невисокі, але мають міцну статуру (кремезні);- риси обличчя досить грубі;
- скелетна мускулатура розвинена добре;
- відзначається гіпертрихоз (посилене оволосіння);
- шкірні покриви на пальцях кистей (частіше) і стоп (рідше) потовщені і натягнуті;
- розміри печінки і селезінки, як правило, не змінені.
 хворі невисокі, але мають міцну статуру (кремезні);
хворі невисокі, але мають міцну статуру (кремезні);Крім того, у таких пацієнтів визначаються порушення, які можуть розцінюватися як окремі захворювання:
- пахові або пупкові грижі;
- синдром зап'ястного каналу - він розвивається через вираженого здавлювання серединного нерва. Виявляється атрофією (недорозвиненням) м'язів тенара (піднесення в області великого пальця кисті) і парестезіями (порушенням чутливості) в області 3, 4 і 5 пальців;
- аортальнийстеноз - звуження аорти;
- недостатність клапанів аорти;
- пігментна дистрофія сітківки;
- глаукома;
- помутніння рогівки.
Інтелект пацієнтів з діагнозом мукополисахаридоза типу I-S залишається в нормі, на відміну від хворих з мукополісахаридозом типу IH.
З інструментальних методів найбільш інформативною є рентгенографія. При цьому визначаються зміни, які схожі на зміни при мукополісахарідозов типу IH, але вони менш виражені.
Мукополісахарідоз типу II
Цей різновид мукополисахаридоза ще називається хворобою Хантера. Патологією найчастіше страждають хлопчики віком 2-3 років.
За клінічними проявами захворювання схоже на мукополісахарідоз типу IH - при цьому спостерігаються:
- скароцефалія;
- грубі риси обличчя;
- низький тембр голосу;
- утруднення дихання - з'являється через деформацію кісток лицьового скелета.
При подальшому прогресуванні захворювання приєднуються наступні ознаки:
- знижується інтелект;
- порушується координація рухів;
- настрій у такого пацієнта нестійке - наголошується так звана емоційна лабільність - від нападів сміху до плачу. Такі різкі перепади настрою можуть з'явитися в будь-який час.
При прогресуванні мукополисахаридоза типу II з'являється і наростає безпричинна агресивність.
На відміну від мукополисахаридоза типу IH, порушення з боку хребта у вигляді кіфозу не є характерними, симптом "котячої спини" не відзначається.
При фізикальному обстеженні визначається наступне:
- виявляється незначне збільшення селезінки і печінки;
- на шкірі спини визначаються специфічні вузлики.
При інструментальному обстеженні виявляються:
- помутніння рогівки, яке прогресує;
- наростаюча приглухуватість.
З усіх інструментальних методів діагностики найбільш інформативною є рентгенографія різних фрагментів опорно-рухового апарату - черепа, рудної клітки, таза і так далі. При цьому рентгенологічні ознаки ті ж, що і при мукополісахарідозов типу I-S.
Пацієнти з таким діагнозом схильні до розвитку захворювань дихальної системи:
- трахеїти;
- бронхітів;
- пневмоній.
Слід врахувати, що мукополісахарідоз типу II може розвиватися у вигляді двох варіантів:
- несприятливого (варіант А);
- сприятливого (варіант В).
 Несприятливий варіант проявляється яскравою клінічною картиною, при цьому зазначаються істотні порушення інтелекту. Летальний результат може наступити в підлітковому періоді життя.
Несприятливий варіант проявляється яскравою клінічною картиною, при цьому зазначаються істотні порушення інтелекту. Летальний результат може наступити в підлітковому періоді життя.
Симптоматика при сприятливому варіанті невиражена, вище описані порушення можуть супроводжуватися незначною розумовою відсталістю. Такі пацієнти, як правило, доживають до 30 років, в ряді випадків - і більше.
Мукополісахарідоз типу III
Мукополісахарідоз типу III описав американський педіатр Санфіліппо, тому патологія отримала ще одну назву - хвороба Санфіліппо.
Захворювання діагностується у 1 дитини на 100-200 тисяч новонароджених.
Діти з мукополісахаридозом типу III в перші роки свого життя розвиваються відповідно до вікових норм, при цьому можуть спостерігатися окремі прояви патології - зокрема, утруднення ковтання і незграбна хода.
Починаючи з 3-5 років, дитина починає відставати від своїх однолітків у фізичному розвитку, при цьому він стає апатичним, байдужим до людей і подій. Типовими є такі ознаки:
- порушення мови;
- огрубіння рис обличчя;
- регулярне нетримання сечі (не тільки нічний) і калу.
У міру дорослішання дитини розумова відсталість наростає і в кінцевому результаті стає основною ознакою цього різновиду мукополисахаридоза.
При фізикальному обстеженні виявляються:
- несуттєве збільшення печінки і селезінки;
- рівномірний посилення оволосіння;
- контрактури;
- затримка росту.
 Порушення з боку органу зору і серцево-судинної системи, які досить часто виявляються при інших формах описуваної патології, для даного типу мукополисахаридоза нехарактерні.
Порушення з боку органу зору і серцево-судинної системи, які досить часто виявляються при інших формах описуваної патології, для даного типу мукополисахаридоза нехарактерні.
Результати рентгенологічного дослідження при описуваної патології або без патологічних змін, або ж схожі на ті, які виявляються при мукополісахарідозов типу I-S, але менш виражені.
Прогноз при даному захворюванні менш сприятливий, ніж при інших типах мукополісахарілоза - такі пацієнти гинуть у віці від 10 до 20 років, причиною летального результату стають інфекційні ускладнення.
Мукополісахарідоз типу IV
Мукополісахарідоз типу IV ще називається хворобою Морков (по імені уругвайського педіатра, який її описав). Патологія зустрічається частіше, ніж інші типи мукополисахаридоза - а саме у 1 дитини на 40 тисяч новонароджених.
Діти з таким захворюванням до 1-3 років розвиваються без особливостей. Але далі з'являються такі порушення:
- дитина істотно відстає в рості від однолітків;
- в процесі розвитку у нього відзначаються вкорочення шиї і тулуба;
- розвиваються різні контрактури;
- потовщується шкіра;
- сила м'язів зменшується;
- слух погіршується;
- риси обличчя стають грубими;
- виникають різні деформації грудної клітини.
Також при мукополісахарідозов типу IV відзначаються порушення, які можуть розцінюватися як окремі патології:
- вальгусна деформація кінцівок - викривлення в напрямку назовні;
- сколіоз (викривлення хребта в бічній площині) або кіфоз;
- пахова грижа;
- пупкова грижа;
- дистрофія рогівки.
Інтелект при такому вигляді мукополисахаридоза збережений.
Діагноз описуваної патології підтверджують за допомогою рентгенологічного дослідження, воно в даному випадку найбільш інформативне. Це такі його види:
- рентгенографія хребта - на знімках визначаються викривлення хребетного стовпа у вигляді кіфозу і сколіозу, а також розширення і сплощення тіл хребців;
- рентгенографія тазу - виявляються множинні деформації тазового кісткового кільця, нерівність його контурів;
- рентгенографія кінцівок - виявляються сплощення головок стегнових кісток, виразне вкорочення кісток передпліччя і деформація кісток стоп.
Хворі з мукополісахаридозом типу IV живуть, як правило, менше 20 років. Летальний результат наступає на тлі ряду супутніх захворювань, які в кінцевому результаті ускладнюються серцево-легеневою недостатністю.
Мукополісахарідоз типу VI
Інша назва мукополисахаридоза типу VI - хвороба Марото-Ламі (за іменами двох французьких лікарів, які описали цю патологію).
Клінічні ознаки багато в чому "перегукуються" з симптоматикою інших видів мукополисахаридоза, але найбільш вираженими є:
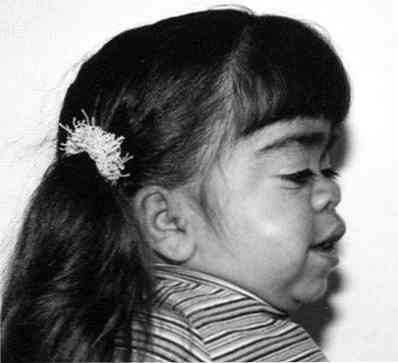 істотне огрубіння рис обличчя;
істотне огрубіння рис обличчя;- помітне відставання в рості;
- вкорочення шиї - іноді здається, що голова пацієнта "сидить" мало не на його плечовому поясі;
- бочкообразная деформація грудної клітини.
 істотне огрубіння рис обличчя;
істотне огрубіння рис обличчя;У таких хворих дуже часто виникають застуди - дана ознака дозволяє запідозрити саме цю, а не іншу форму мукополисахаридоза.
При фізикальному обстеженні виявляється наступне:
- печінка і селезінка часто збільшені;
- можуть діагностуватися пахові грижі.
В інтелектуальному плані дитина розвивається без порушень - у нього збережена здатність мислити і вирішувати інтелектуальні завдання.
Розрізняють два варіанти такого виду мукополисахаридоза:
- класичний;
- мукополісахарідоз зі несуттєвою виразністю клінічних проявів.
Підтвердити діагноз допоможе рентгенологічне обстеження - на знімках визначаються:
- деформація хребців - вони стають Кубовидний або клиноподібними;
- трикутна деформація тазового кільця;
- гіпоплазія (недорозвинення) і деформація головок стегнових кісток;
- вкорочення малогомілкової кісток.
Мукополісахаридози типу VII і VIII
 Мукополісахаридози цих типів зустрічаються рідше, ніж інші різновиди описуваної патології.
Мукополісахаридози цих типів зустрічаються рідше, ніж інші різновиди описуваної патології.
Мукополісахарідоз типу VII клінічно протікає, як хвороба типу III, різниця між ними виявляється тільки в результатах біохімічних досліджень.
Мукополісахарідоз типу VIII по симптоматиці схожий на мукополісахарідоз типу IV. Але відмінність в проявах є - це порушення інтелектуального розвитку, яке не спостерігається при мукополісахарідозов типу IV.
діагностика
При постановці діагнозу мукополисахаридоза грунтуються на скаргах, анамнестичних даних, результатах додаткових методів обстеження - фізикальних, інструментальних, лабораторних. Запідозрити наявність мукополисахаридоза можна за характерними деформацій скелета, які "кидаються" в очі, але визначити тип описуваного захворювання представляється можливим тільки за допомогою додаткових методів обстеження. Головними з таких методів є:
- рентгенографія;
- виявлення глікозаміногліканів в сечі;
- визначення активності ферментів в клітинних культурах.
При клінічних ознаках порушень з боку внутрішніх органів залучаються методи їх обстеження:
- електрокардіографія;
- ультразвукове дослідження печінки та селезінки;
- електроенцефалографія;
- біохімічний аналіз крові
і багато інших.
Диференціальна діагностика
Диференціальну (відмінну) діагностику проводять між різними типами мукополисахаридозов.
ускладнення
Найбільш частими ускладненнями мукополисахаридозов є:
- контрактури;
- серцева недостатність;
- легенева недостатність.
лікування
Терапія мукополисахаридоза, за допомогою якої можна було б усунути хвороба, а не тільки її прояви, не розроблена. Таким пацієнтам проводять симптоматичне лікування - воно може бути:
- консервативне - переливання крові, гормонотерапія, вітамінотерапія і так далі;
- оперативне - ендопротезування головок стегнових кісток, ліквідація гриж, хірургічне втручання для усунення контрактур і виправлення деформацій скелета та інші.
Важливими є:
- лікування інфекцій дихальних шляхів;
- корекція порушень зору і слуху.
профілактика
Так як патологія виникає через генетичних порушень, специфічних методів профілактики не існує. Але ризик генних порушень, які здатні привести до розвитку різних типів мукополисахаридоза, можна знизити, якщо забезпечити жінці нормальні умови протікання вагітності. це:
- відмова від шкідливих звичок;
- дотримання режиму роботи, відпочинку, сну;
- оптимістичний настрій.
прогноз

Прогноз при мукополісахарідозов в цілому несприятливий, це стосується всіх типів описуваного захворювання. В процесі життя і розвитку в тканинах прогресивно накопичуються продукти обміну, які виникають через відсутність тканинних ферментів. Це призводить до патологічних змін з боку практично всіх органів і систем. Якщо патологія запущена, то летальний результат може наступити не тільки через серцево-легеневої, а й полісистемній недостатності.
Консервативна терапія виконує роль підтримуючої - застосування будь-яких медикаментозних засобів веде до тимчасового поліпшення, але не здатне компенсувати брак тканинних ферментів.
Ковтонюк Оксана Володимирівна, медичний оглядач, хірург, лікар-консультант