

Хромосомні патології відносяться до одних з найбільш серйозних поломок в клітинах, за рахунок подвоєння однієї з пар хромосом виникають особливі синдроми, що мають типові зовнішні прояви і зміни метаболізму. Трисомія по одній з пар хромосом (коли замість двох, є три хромосоми) зустрічається найчастіше в трьох варіантах - синдром Дауна, який найбільш поширений і відомий, а також Патау і Едвардса. Свої назви вони отримали за рахунок відкриття і опису їх вченими.
Загальні дані
Синдром Едвардса (або синдром трисомії 18) стоїть на другому місці за зустрічальності трисомій, після синдрому Дауна, і він виникає при порушенні розподілу 18-ї пари хромосом. Патологія характеризується різними порушеннями у формуванні ще внутрішньоутробно систем і органів плода, має зовнішні типові зміни і несприятливі для здоров'я і життя прогнози.
важливоДіти ставляться до категорії важких інвалідів, вимагають до себе особливого ставлення, догляду і турботи батьків і медиків.
 За поширеністю дана хромосомна аномалія вважається досить частою, до 0.01-0.02% дітей, народжених в світі, мають дане хромосомні відхилення, і немає залежності від певної раси чи країни проживання, зустрічається повсюдно. За даними статистики дівчинки страждають частіше хлопчиків в 3-4 рази, хоча аргументованих наукових даних цього феномену, не знайдено. Точна причина формування трисомії у плода не визначена, але виділений ряд певних факторів ризику, які підвищують вірогідність її розвитку.
За поширеністю дана хромосомна аномалія вважається досить частою, до 0.01-0.02% дітей, народжених в світі, мають дане хромосомні відхилення, і немає залежності від певної раси чи країни проживання, зустрічається повсюдно. За даними статистики дівчинки страждають частіше хлопчиків в 3-4 рази, хоча аргументованих наукових даних цього феномену, не знайдено. Точна причина формування трисомії у плода не визначена, але виділений ряд певних факторів ризику, які підвищують вірогідність її розвитку.
Поряд з іншими хромосомними і генними поломками, на сьогоднішній день синдром Едвардса відносять вродженим і невиліковним патологій, методи особливого догляду і медикаментозна підтримка, реабілітаційні заходи дозволяють певним чином підтримувати життя дітей і формувати деякі успіхи в його розвитку - фізичному і психічному. але в силу різкого різноманітності порушень, які надає зайва хромосома, єдиних методик лікування та догляду немає.
Історія вивчення патології
У медичних трактатах і художній літературі можна знайти поодинокі відомості про хворих даними синдромом ще в давнину, але описаний докладно синдром був на початку минулого століття. Розвиток технологій того часу і раніше не дозволяв детально вивчити дану патологію, тому причин її не знали. Крім того, більшість народжених з цим синдромом дітей гинуло в ранньому віці в силу слабкого розвитку медицини і своєчасного надання їм допомоги. Причина синдрому - трисомія по 18-ій парі хромосом була виявлена тільки в 1960 році Д. Едвардсом, і його ім'ям і була названа нова патологія. За даними медичної статистики зустрічальність патології в середньому становить 1 випадок на 3000 зачать, але до пологів залишається набагато менше плодів. Це пояснюється тим, що подібна аномалія нерідко на самому початку закінчується викиднем або загибеллю плоду внутрішньоутробно. Але нерідко при таких викидні ніяких хромосомних досліджень не проводиться.
Що таке трисомия
Якщо говорити з точки зору медицини, трисомія є варіантом хромосомних мутацій, коли клітини пацієнта мають не 46 хромосом зібраних в гомологічні 23 пари, одна пара з яких визначає статеві відмінності, а виявляється 47 хромосом.
Сьогодні лікарі виділяють три типи трисомії, які відносяться до нелетальної мутацій - це синдром Дауна (страждає 21-а пара), синдром Патау (страждає 13-а пара) і синдром Едвардса (поразка виявляється по 18-ій парі). Всі інші варіанти трисомії спочатку несумісні з життям, такі зародки відразу ж гинуть і вагітність не розвивається. Але народження дітей з цими трьома трисомиями - це завжди проблеми їх здоров'я, фізичного розвитку та інтелекту, психічних функцій.
Причини розвитку синдрому Едвардса
Як і дві інших патології, зазначені вище, синдром Едвардса відносять до генетичних патологій, при яких виявляється додаткова хромосома ваутосомах (НЕ статеві хромосоми). У здорової людини 46 хромосом - 22 пари - аутосоми (нестатеві), і одна пара - статеві, що визначають - дівчинка це чи хлопчик. У хромосомах компактно упаковані нитки ДНК, в якій записана вся генетична інформація про організм, починаючи з процесу його зачаття і до смерті. Вся програма життя записана саме в нитках ДНК.
При синдромі Едвардса організм страждає не від генних дефектів, а хромосомних, виникає дефект в розподілі статевої клітини, яка потім дає дитині життя, і в ній залишається зайва хромосома. При класичному варіанті формування синдрому Едвардса є подвоєння 18-ї хромосоми, і народитися можуть з подібним синдромом як хлопчик, так і дівчинка. У всіх клітинах таку дитину існує надлишок генетичної інформації. Подібна надмірність ДНК призводить до цілого ряду порушень, які призводять як до зовнішніх дефектів, так і проблемам в роботі систем і органів. Через наявність третьої, зайвої хромосоми і виникло наукова назва - трисомія.
Типи синдрому Едвардса: як залежить тяжкість проявів
Виходячи з того, якого роду хромосомний дефект є при синдромі, виділяють три типи перебігу:
- Повна трисомія по 18-ій парі.
- Часткова трисомія по 18-ій парі
- Мозаїчна форма патології.
при повній формі (Вона ж відноситься до класичного синдрому) передбачається, що абсолютно у всіх клітинах є додаткова хромосома. Подібний варіант найбільш важкий і зустрічається найбільш часто, прогнози при ньому найсприятливіші.
 часткова трисомия відноситься до рідкісних варіантів патології, виникає в 3% випадків, при такій проблемі міститься не повна третя хромосома, а тільки її шматок. Подібний дефект може утворюватися в результаті проблем з поділом клітин (особливо їх хромосом), що виникає вкрай рідко. Може бути і варіант того, що зайва частина 18-ї хромосоми кріпиться до окремих молекул ДНК, приводячи до їх подовження і зміни. У таких клітинах потім залишається дві 18-их хромосоми і частина зайвого матеріалу. Тоді вроджений синдром буде не таким важким, так як в клітинах з'являється додаткова інформація не всієї інформації, а тільки її частини. Прогноз при такому стані хоча і несприятливий, але перебіг синдрому набагато легше, ніж при повній формі.
часткова трисомия відноситься до рідкісних варіантів патології, виникає в 3% випадків, при такій проблемі міститься не повна третя хромосома, а тільки її шматок. Подібний дефект може утворюватися в результаті проблем з поділом клітин (особливо їх хромосом), що виникає вкрай рідко. Може бути і варіант того, що зайва частина 18-ї хромосоми кріпиться до окремих молекул ДНК, приводячи до їх подовження і зміни. У таких клітинах потім залишається дві 18-их хромосоми і частина зайвого матеріалу. Тоді вроджений синдром буде не таким важким, так як в клітинах з'являється додаткова інформація не всієї інформації, а тільки її частини. Прогноз при такому стані хоча і несприятливий, але перебіг синдрому набагато легше, ніж при повній формі.
розвиток мозаїчної форми проявляється приблизно у 5-8% дітей з подібною аномалією. Але за механізмами подібний тип синдрому відрізняється. Цікаво, що подібна форма розвивається після того, як сперматозоїд зливається з яйцеклітиною, і обидві вийшли клітини спочатку мають нормальним набором хромосом - 46ХХ або 46XY. Але при перших же розподілах в утвореній зиготі утворився збій і одна з клітин придбала в силу цього додаткову хромосому. В результаті зародок придбав клітини зі звичайними хромосомами - їх 46, інша ж частина з них має зайву хромосому - то є 47. Кількість клітин із зайвою хромосомою при таких проблемах не перевищить 50%.
Зверніть увагуЧим вище кількість дефектних клітин у зародка і дитини, тим гірше прогнози, і залежить їх обсяг від того часу, коли стався збій при розподілі. Чим пізніше це сталося, тим менший обсяг дефектних клітин має крихітка, і знижена кількість проблем розвитку.
Дана форма отримала назву мозаїчної в силу того, що нормальні клітини перемішані з дефектними. Атипові клітини із зайвою хромосомою розкидані хаотично по всьому організму, і немає способу, щоб можна було їх яким-небудь чином видалити. Ця форма протікає дещо легше, тривалість життя вище.
Основа патології: в чому провина генів?
Всього одна хромосома у дитини має багато серйозних і важких наслідків для здоров'я і життя. Це пов'язано з тим, що у всіх клітинах є універсальна система зчитування інформації, і при діленні клітин - подвоєння тієї інформації, що запрограмована в молекулах ДНК. Якщо є дефект хоча б одного гена - це вже відбивається на організмі у вигляді будь-якої хвороби або проблеми, а якщо є цілий шматок зайвої хромосоми або навіть ціла хромосома - це множинні вади розвитку і проблеми здоров'я.
На 18-ій хромосомі міститься до 2.5% всієї генетичної інформації про роботу організму, і порушення, які відтворюються за рахунок синдрому - дуже значні. Зайві гени на дефектної хромосомі дають початок синтезу зайвих білків, що призводить до аномального розвитку і функціонуванню цілого ряду органів і систем дитини. При синдромі Едвардса піддаються ураженню скелет, певні зони нервової системи, сечостатева система і серцево-судинна, вони мають як аномалії в будові, так і потім - у міру розвитку плоду - в функціонуванні.
Відповідно - основна і єдина причина подібного синдрому - освіту в статевій клітині матері або батька зайвої хромосоми. При виробництві статевих клітин відбувається не традиційне розподіл з подальшим подвоєнням 23 пар хромосом до 46 штук (для кожної ідентична копія в пару), а всього 23 штук. Якщо в процесі ділення 18-я пара з якихось причин правильно не розділені, в статеву клітину піде відразу дві хромосоми, і буде в ній не 23 штуки, а 24-ре. Якщо така клітина дасть початок нового життя (будь то татів сперматозоїд або мамина яйцеклітина), це призведе до появи дитини з синдромом Едвардса.
Фактори ризику синдрому Едвардса
Щоб статеві клітини батьків неправильно ділилися, і утворилася та, що має надлишкову набір хромосом, необхідний різні негативні впливи, як ззовні, так і всередині організму. Провідними з них можна вважати:
- вік матері і батька до моменту зачаття. Є реальні докази вікового фактора на ймовірність розвитку трисомії. У міру віку число аномальних клітин в репродуктивній системі збільшується, що пов'язано з ослабленням контролю з боку організму за процесами поділу. Ризик народження дітей з хромосомними патологіями після 30 років збільшується в 2-3 рази в порівнянні з молодим віком, а після 40 років шанси збільшуються ще в 6-8 разів. Залежність від віку батька не так очевидна, але теж цілком можливі хромосомні дефекти.
- наявність шкідливих звичок, особливо якщо це нікотинова й алкогольна залежність. Звичні інтоксикації порушують процеси ділення клітин, в тому числі і в області статевої сфери, що призводить збільшення ризику дефектних сперміїв і яйцеклітин в кілька разів. Ще небезпечніше прийом наркотичних препаратів і токсикоманії, вони ще сильніше порушують процеси клітинного ділення.
- прийом деяких лікарських засобів, впливають на процеси клітинного ділення, перевищення дозувань і тривалості прийому, несприятливі поєднання препаратів при лікуванні, що формують токсичні їх концентрації.
- інфекції соматичні і статеві, які призводять до ураження репродуктивних органів і клітин, формують хронічний перебіг, впливають на процеси ділення (цитомегалія, герпес, папілома і інші).
- професійні шкідливості, опромінення різного типу - іонізуючі, магнітні і багато інших. Вони впливають на процес ділення клітин, мають шкідливою дією відносно ДНК, призводять до утворення в клітинах вільних радикалів і мутацій.
Остаточні причини та всі фактори ризику, які призводять до утворення клітин з трисомія, можуть бути з'ясовані в міру відкриттів молекулярної біології і генетики. Всі перераховані фактори тільки збільшують ризики, але не говорять про те, що обов'язково народиться така дитина.
Не виключена і схильність до утворення трисомії в статевих клітинах, що доводиться тим фактором, що при народженні в сім'ї однієї дитини з синдромом Едвардса, шанси на народження ще одного малюка підвищуються в 200 разів у порівнянні з середньостатистичними - з 0.04% до 2-3% і більше.
Характеристики синдрому Едвардса в період новонародженості
Хоча сьогодні існує можливість діагностики синдрому до народження, часто патологію виявляють уже при пологах. Новонароджений з подібною аномалією хромосом має типові і яскраво виражені пороки в розвитку, їх наявність може допомогти в постановці діагнозу навіть без додаткових досліджень хромосом - генетичного аналізу (але його проводять в будь-якому випадку, без цих даних діагноз не остаточний).
При народженні виявляються:
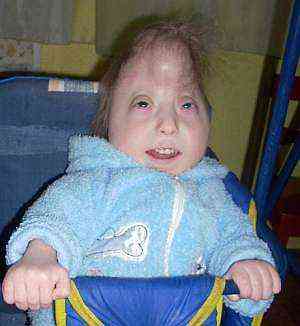 аномалії в будові форми черепа. Скелет при подібному синдромі страждає найбільш виражено - це проявляється в зміні насамперед форми черепа з утворенням доліхоцефалією (різко витягнутий, вузький череп - "баштовий"). Подібна аномалія не типова тільки для цієї патології, але вказує на наявні відхилення в розвитку. Найбільш виражено зміна співвідношень за даними вимірів в області тім'яних кісток і довжини від перенісся до потиличних горбів. Важчій і вираженою аномалією черепа може стати мікроцефалія, непропорційно маленький розмір мозкового черепа щодо тіла.
аномалії в будові форми черепа. Скелет при подібному синдромі страждає найбільш виражено - це проявляється в зміні насамперед форми черепа з утворенням доліхоцефалією (різко витягнутий, вузький череп - "баштовий"). Подібна аномалія не типова тільки для цієї патології, але вказує на наявні відхилення в розвитку. Найбільш виражено зміна співвідношень за даними вимірів в області тім'яних кісток і довжини від перенісся до потиличних горбів. Важчій і вираженою аномалією черепа може стати мікроцефалія, непропорційно маленький розмір мозкового черепа щодо тіла.- специфічно змінена форма вушної раковини. Аномальне будова вушних раковин у новонароджених виражена сильно і спостерігаються у 95% малюків, особливо при повному типі синдрому. Розташовані вушні раковини набагато нижче звичайного, щодо очниць вони різко опущені, хрящі опуклі і немає чітко сформованих завитків. Мочка і козелок в зародковому стані або повністю відсутні, слуховий прохід різко звужується і у чверті дітей може бути відсутнім зовсім.
- аномальне будова неба з нижньою щелепою (формування мікрогнатії). У новонароджених, коли підозрюють синдром Едвардса, немає повного зрощення піднебінних відростків в зоні верхньої щелепи, що утворюють поздовжню щілину. Можуть бути варіанти з тотальним незарощення тільки м'якого піднебіння або частковим його зрощенням, іноді з переходом на губи. При повній формі пороку незарощення може бути двостороннє, верхня губа розщеплена з двох сторін, дає початок патологічним ущелинах в щелепах і небі, що не дає дитині повністю закривати рот. У разі важких патологій можливі сполучення між ротоглотки і носової порожнин, і в тому випадку, якщо рот закритий. Частота таких аномалій досягає 20-25% і вище.
- Можуть виникати й інші проблеми - готичне небо (Надмірно високий звід кісток) і зміни в будові нижньої щелепи. Їх реєструють приблизно у 70% дітей. типова микрогнатия - сильна втягнути підборіддя і нижньої щелепи назад, що не дає немовлятам повністю закривати рот і слина підтікає з рота. При подібній аномалії виникають труднощі з годуванням, особливо прикладанням до грудей.
- деформація стоп з утворенням "стопи-качалки", характерні аномалії довжини пальців кінцівок. Зміна стопи типово для 75% і більше дітей на тлі даного синдрому, виникає зміна положення кісточок - таранної, п'яткової або човноподібної, що призводить до формування сильно видатної п'яти на тлі відсутності склепіння стопи. Він може бути вигнутий вперед, надаючи стопах вид ніжок від крісла-качалки. Також виявляються і диспропорції в довжині пальців на ногах, особливо великого. Він коротше другого пальця на нозі, що видно при розпрямлених пальчиках.
- кисті в флексорного положенні і зрощення між пальцями, особливі дерматогліфічні малюнки. синдактилія (Цей термін позначає зрощення пальців) виявляють у 45% випадків, зазвичай це пальці на ногах, але можуть порушуватися і руки. При легкому ступені шкіра створює перетинки, в важких можуть утворюватися кісткові перемички. Також можуть бути флексорние аномалії кистей - становище пальців в аномальному, постійно напруженому положенні, прикриваючи мізинцем і великим пальцем всі інші, щільно притиснуті до долоні. Типові подібні розлади для 90% дітей з синдромом.
- Крім усього іншого, при трисомії типові особливі візерунки на пальцях і долонях з особливою формою шкірних складок. При розвитку цього синдрому відхилення типові для 60% малюків. Це дуже часті дуги на подушечках, відсутність шкірної складки між крайньою і передостанній фалангами, розвиток поперечний борозенки - вона має термін "мавпяча" лінія.
- пороки геніталій. У хлопчиків це недорозвинення пеніса і мошонки, у дівчаток збільшення розмірів клітора. Аномалії поєднуються зазвичай з вадами сечостатевої системи, але зовні інші ознаки не помітити.
 аномалії в будові форми черепа. Скелет при подібному синдромі страждає найбільш виражено - це проявляється в зміні насамперед форми черепа з утворенням доліхоцефалією (різко витягнутий, вузький череп - "баштовий"). Подібна аномалія не типова тільки для цієї патології, але вказує на наявні відхилення в розвитку. Найбільш виражено зміна співвідношень за даними вимірів в області тім'яних кісток і довжини від перенісся до потиличних горбів. Важчій і вираженою аномалією черепа може стати мікроцефалія, непропорційно маленький розмір мозкового черепа щодо тіла.
аномалії в будові форми черепа. Скелет при подібному синдромі страждає найбільш виражено - це проявляється в зміні насамперед форми черепа з утворенням доліхоцефалією (різко витягнутий, вузький череп - "баштовий"). Подібна аномалія не типова тільки для цієї патології, але вказує на наявні відхилення в розвитку. Найбільш виражено зміна співвідношень за даними вимірів в області тім'яних кісток і довжини від перенісся до потиличних горбів. Важчій і вираженою аномалією черепа може стати мікроцефалія, непропорційно маленький розмір мозкового черепа щодо тіла.Крім цих проявів є і більш дрібні ознаки і аномалії розвитку дітей, які можуть в попередньому діагностуванні синдрому. Важливо відзначити, що один з перерахованих ознак і навіть два ще не говорять про діагноз, при синдромі пороки множинні і мають різні поєднання.
важливоБільшість з описаних проявів можуть зустрічатися як окремі види і діагнозу не підтверджують, вони можуть бути ізольованим варіантом відхилень в перебігу вагітності або проявом інших патологій. Діагноз тільки за зовнішніми порокам ставити не можна!
Більш того, на тлі неповного або музичного типів синдрому зовнішніх ознак може бути трохи - один-два, при цьому є серйозні вади внутрішніх органів з частими відхиленнями в хромосомах. Саме вони і визначають подальші прогнози на життя і перебіг синдрому.
Розвиток дітей: особливості синдрому Едвардса
Тільки зовнішнім виглядом при появі на світ і проявами вад в розвитку, аномалія Едвардса не обмежується, у міру зростання і розвитку можливі прояви все нових і більш важких відхилень. Перші з них можна виявити через кілька тижнів з моменту народження. Нерідко по ним вперше можна запідозрити діагноз при мозаїчному і неповному типі патології, якщо зовнішніх дефектів мало або практично немає. Описані вище пороки і зовнішні зміни стають у міру зростання більш різкими і вираженими, поступово додаються нові проблеми і аномалії, які при народженні не були помітні. Так, сюди варто віднести:
- Різке і виражене відставання в рівні фізичного розвитку. При народженні маса таких дітей не перевищує 2-2.5 кг, і причиною тому є хромосомні дефекти, які не дають повноцінно розвиватися системам і органам. У міру розвитку показники росту і маси тіла по місяцях істотно відрізняються від норм, також слабо прибувають і окружності грудей. Розміри голови можуть прибувати але нормі або вище неї, що може вказувати на скупчення всередині порожнини черепа надлишку рідини, формуючи гідроцефалію.
- Проблеми зі стопами. У міру зростання формується клишоногість в силу аномалій будови кісток і зв'язок, м'язового гіпертонусу. Страждає і контроль за розвитком стопи і тонусом від нервової системи. Це загрожує тим, що діти пізно починають ходити або не можуть цього робити взагалі в силу загального важкого стану. Зовні клишоногість проявляється у вираженій деформації стопи і ненормальною її установці в спокої.
- Порушення м'язового тонусу. При народженні гіпертонус типовий для кистей, формуючи їх флексорного положення, але в міру розвитку поширюється на сусідні груп м'язів, приводячи до химерним позам. Але частіше тонус м'язів знижений, діти мляві і атонічние, кінцівки важать батогами. Можуть також бути стану дистонії з різким скороченням окремих м'язових груп на тлі гіпотонії інших. При цьому руки можуть бути зігнуті, а ноги - перерозгинати. Це призводить до порушення координації і хаотичності рухів, вивихів і ненормальному становищу кінцівок.
- Атипові реакції нервової системи, проблеми емоційності. Деякі відділи мозку у дітей з синдромом Едвардса недорозвинені, в зв'язку з чим формуються проблеми емоційності і загальмованості, неадекватні реакції на навколишнє. Це пов'язано зазвичай з мозолясті тілом і мозочковими гіпоплазії, що формує також відставання розумового розвитку. Зовні це проявляється в відсутньому погляді, неемоційне і неможливості встановлення контакту, відсутності стеження за об'єктами. Діти не реагують на звуки в силу проблем з вухами і нервовою системою, подібні аномалії виявляють у перші ж місяці життя.
В цілому, в силу важких і комбінованих уражень діти не доживають до періоду повноліття. Частина їх при мозаїчній формі може досягати віку підлітків з глибокими ступенями імбецильності і зовнішніми проявами вад.
Методи діагностики синдрому Едвардса
На сьогоднішній день існує три типи діагностик синдрому, які принципово відрізняються один від одного. З огляду на той факт, що патологія невиліковна і летальна в ранньому віці, важливо звертати увагу на пренатальну діагностику, яка не допускає народження дітей з даними аномаліями. Корисними будуть і консультації лікарів-генетиків в сім'ях, де мали місце випадки хромосомних патологій. Можна провести:
- Діагностику статевих клітин ще до зачаття. На жаль, подібний метод можна застосовувати ще дуже обмежено і лише в протоколах ЕКО при попередньому вивченні статевих клітин. Якщо ж це природне зачаття, лікарі можуть прогнозувати більш високі ризики народження таких дітей, але це не більше, ніж прогнози. Точно дізнатися - чи народиться така дитина чи ні - неможливо. Досліджується сімейний анамнез з урахуванням всіх наявних захворювань, а також оцінюються фактори ризику, а також проводиться генетичний аналіз обох батьків із застосуванням складної апаратури та аналізів. За рахунок визначення каріотипу батьків можуть робитися певні висновки.
- Виявлення синдрому в період гестації. При розвитку плода існує кілька методів прямого або непрямого підтвердження синдрому Едвардса, спочатку для цих цілей застосовують УЗД-скринінг і аналізи крові з вивченням особливих показників, а при високих ризиках проводяться вже інвазивні процедури - амніоцентез, або кордоцентез і біопсія ворсинок хоріона. Проводять скрининги в строго визначені терміни, коли зміна даних УЗД і показників крові буде найбільш значущим.
Отримання безпосередньо клітин плоду або його крові дозволяє провести дослідження його хромосом і підтвердити або спростувати у нього синдром Едвардса. Це допомагає в ухваленні рішення про пролонгацію або переривання вагітності за медичними показаннями.
- Підтвердження синдрому після народження. Його проводять за результатами зовнішніх даних в комбінації з рядом генетичних аналізів, які б підтверджували наявність трисомії по 18-ій парі хромосом. Крім цього важливо оцінити стан дитини і дати прогнози щодо його життя і стану здоров'я, щоб зрозуміти, який обсяг допомоги йому потрібно для виживання. Без медичної допомоги при подібному синдромі діти можуть швидко гинути поле народження в найближчі тижні. Для виявлення вад у внутрішніх органах проводять УЗД і ЕХО-КГ, рентгенографію і ЕЕГ, цілий ряд аналізів крові і сечі, додаткові дослідження аж до МРТ і КТ.
Лікування та прогнози при синдромі Едвардса
На жаль, даний синдром на сьогоднішній день невиліковний, і лікарі можуть допомогти дитині тільки краще справлятися з різними проблемами за рахунок оперативних втручань і консервативної терапії. Зазвичай діти доживають до декількох місяців або року, тільки при частковій або мозаїчній формі можуть прожити триваліше. Загибель настає від множинних вад розвитку та аномалій в роботі органів, а також приєднання супутніх ускладнень. Радикально виправити дефекти хромосом в клітинах тіла дитини на сьогоднішньому етапі розвитку медицини неможливо.
Парецкі Алена, педіатр, медичний оглядач